CE сертификат медицинского изделия
CE сертификат медицинского изделия
Единый рынок Европейского союза является одним из самых перспективных для реализации медицинских изделий. В статье мы рассказываем как получить разрешительную документацию для свободной продажи изделий, произведённых за пределами ЕС.
Что такое CE сертификат и CE маркировка медицинского изделия
В переводе с французского сокращение CE означает «европейское соответствие» – conformité européenne. СЕ маркировка (или CE марка) ‒ символ, означающий, что медицинское изделие соответствует регуляторным требованиям Европейского Союза. Регуляторные требования изложены в европейских регламентах:
Медицинские изделия, которые не соответствуют этим требованиям, нельзя импортировать в Европейский Союз и допускать в обращение на его территории.
Соответствие регуляторным требованиям может подтверждаться:
Декларация соответствия для медицинских изделий I класса (нестерильных и не обладающих измерительной функцией) оформляется самим производителем (самодекларирование).
Для остальных изделий для легального обращения не территории ЕС необходимо оформить CE сертификат при участии нотифицированного органа Европейского Союза.
CE сертификат содержит следующие сведения:
Преимущества и актуальность CE сертификации
В последнее время процедура регистрации медицинских изделий в РФ и ЕАЭС стала значительно сложнее и дороже для производителей и импортеров. Только за последний год стоимость испытаний увеличилась в 2‒3 раза.
В то же время значительно сократилось количество лабораторий готовых провести испытания. Это связано как с ужесточением требований к самим лабораториям (со стороны Росаккредитации), так и к результатам испытаний (со стороны экспертных организаций Росздравнадзора). Все это приводит к существенному росту издержек и увеличению сроков проведения испытаний.
Одновременно с этим постоянно меняется законодательство, регулирующее обращение медицинских изделий. Так обещанный полноценный переход на правила ЕАЭС в очередной раз откладывается, при этом национальная процедура по правилам РФ временно приостановлена.
Хотя принято предварительное решение о ее продлении до конца 2022 года, окончательные сроки возобновления национальной процедуры пока не ясны.
В этой связи альтернативой получения регистрационного удостоверения в РФ или ЕАЭС может стать получение СЕ сертификата на медицинское изделие.
Это откроет для производителей и импортеров следующие преимущества:
Этапы CE сертификации медицинского изделия
Процедура получения СЕ сертификата определяется следующими документами в зависимости от типа медицинского изделия:
Разберем основные этапы регистрации медицинского изделия в Европе на схеме с пояснениями.
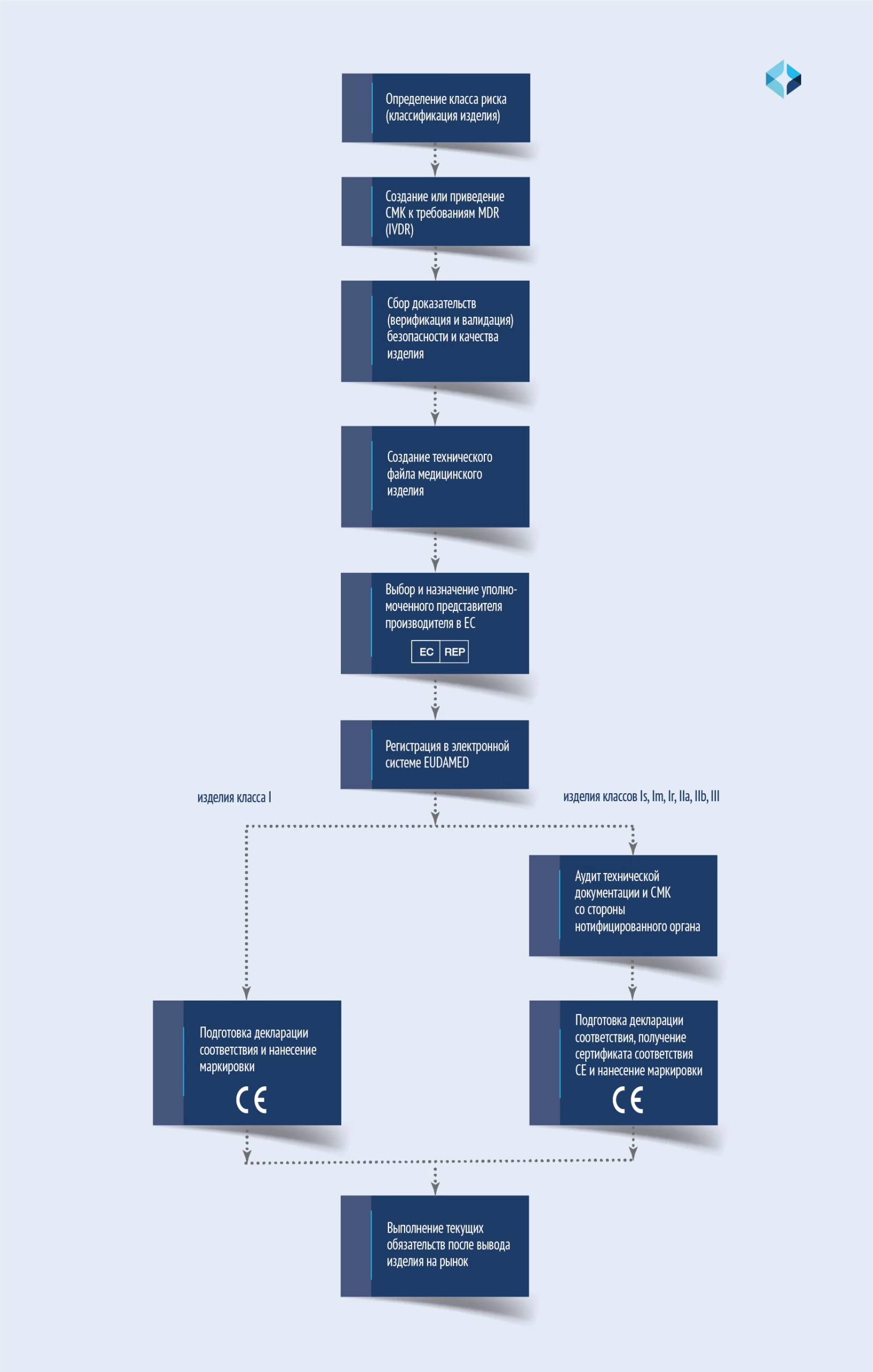
Как видно из схемы, для медицинских изделий I класса (нестерильных и не обладающих измерительной функцией) процедура CE сертификации значительно упрощена. Она не требует участия нотифицированного органа. Поэтому процесс называется самодекларированием. Однако изделие также должно полностью соответствовать требованиям Регламента.
Производитель также обязан внедрить и поддерживать адекватную систему менеджмента качества, хранить и актуализировать технический файл, иметь уполномоченного представителя и своевременно передавать сведения в EUDAMED. Только на этих условиях производитель вправе декларировать соответствие требованиям Регламента, выпускать декларацию соответствия и наносить CE маркировку на изделие.
Степень участия нотифицированного органа в сертификации остальных изделий I класса:
Для изделий классов II и III участие нотифицированной организации в регуляторном процессе значительно возрастает при увеличении класса, поскольку увеличиваются риски.
Европейский представитель производителя медицинского изделия
В соответствии с требованиями регламентов MDR 2017/745 и IVDR 2017/746, у каждого производителя, не являющегося членом Европейского союза, если он планирует продавать свои изделия на территории ЕС, должен быть уполномоченный представитель (European Authorized Representative). Это относится ко всем изделиям вне зависимости от их класса.
Уполномоченный представитель ‒ это юридическое лицо, учрежденное в ЕС, которое получило письменное право действовать от имени производителя, который осуществляет производство изделий за пределами Европейского союза. Задачи и обязательства, которые берет на себя представитель, определяются соответствующими регламентами.
Основные функции и обязанности уполномоченного представителя в ЕС:
Задачи, порученные производителем европейскому уполномоченному представителю, должны быть четко обозначены и закреплены в письменной форме. Нельзя поручать представителю задачи, выполнить которые может только производитель. Например, обеспечение соответствия производственного процесса документации производителя, разработку и актуализацию этой документации.
Строгость требований к уполномоченному представителю в ЕС зависит от класса риска изделий. Поскольку авторизованный представитель должен контролировать соответствие продукта, производителя и его СМК требованиям регламентов ЕС, компетентность самого представителя должна позволять ему работать с медицинскими изделиями в строгом соответствии с требованиями нормативных документов!
На маркировке медицинского изделия сведения об уполномоченном представителе в ЕС обозначаются с помощью символа EC REP (см. рисунок).

На маркировке для европейского уполномоченного представителя указываются его наименование, адрес. Дополнительно можно указать контактный телефон и адрес электронной почты.
Среди наших официальных партнеров есть организации, выступающие уполномоченным представителем в Евросоюзе для производителей медицинских изделий нерезидентов ЕС.
Что такое нотифицированный орган и его функции
Нотифицированные органы – это организации, уполномоченные Европейской комиссией проводить оценку соответствия в ЕС или третьих странах. Каждый орган должен пройти процедуру официальной аккредитации и должен подтвердить свою компетентность в той или иной области. Для оценки соответствия медицинских изделий нотифицированный орган должен продемонстрировать компетентность в отношении MDR 2017/745 и/или IVDR 2017/746 (для изделий in vitro). Перечень аккредитованных нотифицированных органов представлен на сайте Европейской Комиссии в разделе «NANDO нотифицированные органы».
Для каждого Регламента есть отдельные списки нотифицированных органов, которые могут оказывать услуги по аудиту, инспекции и сертификации. По Регламенту MDR 2017/745 на сегодня таких органов не более 30, а по IVDR 2017/746 – меньше 10 (см. рисунки).

Результаты поиска нотифицированных органов (Регламент MDR 2017/745)

Результаты поиска нотифицированных органов (Регламент IVDR 2017/746)
Сведения о нотифицированном органе, который принимал участие в оценке соответствия изделия, приводится на его упаковке и отображается под символом CE в виде четырехзначного номера (NB number).
Что такое технический файл медицинского изделия
Согласно законодательству ЕС для каждого медицинского изделия должен быть разработан технический файл (technical file). Он содержит требования при проектировании, производстве, обращении, эксплуатации и утилизации медицинского изделия. Ответственность за сведения, содержащиеся в техническом файле, возлагается как на производителя, так и на уполномоченного представителя.
Минимально технический файл должен содержать следующие разделы:
Напоминаем, что для изделий IIa, IIb и III классов риска, а также изделий I класса стерильных (Is), многоразовых хирургических инструментов (Ir), изделий с измерительной функцией (Im) осуществляется аудит технического файла со стороны нотифицированного органа!
На практике детальные требования к формату и составу технического файла устанавливаются конкретным нотифицированным органом.
Минимальный срок хранения документов технического файла составляет 10 лет с даты окончания производства.
Полные требования к содержанию технического файла приведены в Приложении II к EU MDR 2017/745 и в Приложении II к EU IVDR 2017/746 в отношении изделий для диагностики in vitro.
Испытания медицинских изделий для СЕ сертификации
В процессе подготовки технического файла производитель должен провести все необходимые испытания медицинского изделия. Целью их проведения является подтверждение соответствия требованиям MDR 2017/745, MDR 2017/746 и применимым к изделиям стандартам.
Условно испытания можно разделить на следующие группы:
В составе технического файла результаты испытаний приводятся в разделе верификация и валидация изделия (Product verification and validation).
Технические испытания ‒ испытания, проводимые с целью определения соответствия характеристик (свойств) изделия требованиям нормативной документации, технической и эксплуатационной документации производителя. К ним можно отнести: определение габаритов, массы, жесткости, испытания функциональные характеристик, проверку устойчивости к климатическим и механическим воздействиям, подтверждение заявленного срока годности/эксплуатации и т.д.
Испытания на биосовместимость – подтверждают безопасность применяемых при производстве медицинского изделия материалов при их контакте (прямом или опосредованном) с телом человека.
Доклинические испытания – проводятся на животных с целью получения данных об эксплуатации медицинского изделия в живом организме до применения на человеке. В силу специфики медицинских изделий далеко не все могут быть предварительно испытаны на животных.
Клинические испытания – испытания с человеком в качестве субъекта испытания. Данные испытания проводятся не всегда. Для получения CE сертификата необходимы клинические данные, подтверждающие клиническую эффективность и безопасность медицинского изделия.
Для изделий с низким классом риска такие клинические данные могут быть получены из научной литературы, клинического опыта применения эквивалентного изделия. В случае имплантируемых устройств и устройств III класса, клинические испытания с человеком в качестве субъекта являются обязательными (за исключением некоторых четко оговоренных ситуаций).
В подавляющем большинстве случаев СЕ сертификат можно получить без проведения клинических испытаний. Однако в любом случае производителю потребуется подготовить отчет о клинической оценке (Clinical evaluation report), включающий анализ всех доступных для оценки клинических данных.
К специфическим испытаниям относятся те, которые проводятся для отдельных видов изделий. В качестве примера можно привести испытания электромагнитной совместимости.
Где проводить испытания медицинского изделия
СЕ сертификация допускает, что часть испытаний производитель может провести самостоятельно на месте производства медицинского изделия. Однако при прохождении аудита СМК вопросы, связанные с проведением испытаний силами производителя, оцениваются нотифицированным органом. Как правило, производитель может подтвердить соответствие габаритных размеров, массы, иные параметры, которые подлежат выходному контролю в процессе производства. При этом все испытания должны быть проведены по методикам испытаний, признаваемым ЕС. Если применяются иные методики, их применение должно быть обосновано, а сами методики валидированы непосредственно производителем.
Испытания, которые производитель не может провести собственными силами (как правило, испытания на электромагнитную совместимость, стерильность, биосовместимость и т.д.) крайне желательно провести в лабораториях, аккредитованных в международной системе ILAC (по стандарту ISO 17025) или лабораториях, признанных национальными органами аккредитации стран ЕС. Область аккредитации таких лабораторий должна включать требуемые стандарты и методики. Результаты, полученные в таких лабораториях, принимаются нотифицированными органами без нареканий.
Доклинические испытания должны быть проведены в исследовательских центрах, соблюдающих требования стандартов и рекомендаций по проведению испытаний с использованием животных. Гуманность и отсутствие необоснованных страданий должны быть приоритетны в процессе проведения испытаний.
Клинические испытания должны быть проведены только после получения заключения этического комитета об одобрении клинических испытаний в тех медицинских учреждениях, в которых имеется опыт применения изделий аналогичных тем, которые планируется испытать.
Что такое система EUDAMED и ее функции
Eudamed ‒ это единая европейская информационная система для медицинских изделий и экономических операторов (субъектов обращения), обеспечивающих вывод и обращение медицинских изделий на рынке ЕС. Регистрация в системе Eudamed является обязательной и определяется требованиями регламентов ЕС MDR 2017/745 и ЕС IVDR 2017/746.
Система состоит из шести взаимосвязанных модулей:
В каждый из указанных модулей вводится соответствующая информация на официальном сайте EUDAMED. Эта информация является общедоступной, что повышает прозрачность рынка медицинских изделий ЕС.
Кто должен регистрироваться в EUDAMED
Регистрация в EUDAMED является обязательной для:
Важно отметить, что регистрироваться должен любой производитель, независимо от того, находится ли он в Европейском союзе или нет.
Если производитель не является резидентом ЕС, к моменту его регистрации в EUDAMED в систему должны быть внесены сведения об уполномоченном представителе производителя на территории ЕС.
В случае выполнения экономическим оператором нескольких ролей, то он должен регистрироваться в системе в каждой из них.
Для получения регистрационного номера (Single Registration Number, SRN) российскому производителю необходимо направить заявку в адрес своего уполномоченного представителя. После этого авторизованный представитель должен одобрить эту заявку.
Только после этого российский производитель получит свой SRN. Если экономический оператор регистрируется в нескольких ролях (например, производитель и уполномоченный представитель), то для каждой из них он должен получить уникальный SRN.
Сроки получения CE сертификата
Сроки зависят от следующих факторов:
Кроме того, Регламент EU MDR ввел дополнительные требования, особенно в отношении систем менеджмента качества. Это делает весь процесс более сложным и трудоемким, а, следовательно, увеличивает сроки оформления сертификата.
По нашему опыту, в среднем сроки получения СЕ сертификата при разработке документов на изделие и СМК с «нуля» следующие:
При наличии части необходимых документов и/или действующего сертификата СМК указанные сроки могут быть существенно сокращены.
Срок действия сертификата CE
CE сертификат обычно действует в течение 3 лет. Для медицинских изделий высокого класса риска этот срок может уменьшаться до 1 года. Максимальный срок может составлять 5 лет. Досрочно действие сертификата CE может прекращаться по результатам неудовлетворительного контрольного аудита со стороны нотифицированного органа.
Стоимость CE сертификата
Общие затраты на сертификацию пропорциональны классу медицинского изделия. Чем выше класс, тем выше риск и сложнее процесс получения сертификата.
Стоимость сертификации медицинского изделия в ЕС складывается из следующих затрат:
Таким образом назвать предварительную стоимость получения СЕ сертификата можно только после изучения имеющихся документов медицинского изделия. На стоимость могут также повлиять пожелания заказчика относительно выбора нотифицированного и сертификационного органов.
Свяжитесь с нами и сообщите, на какое медицинское изделие вы планируете оформить CE сертификат, и мы бесплатно рассчитаем стоимость. Просто позвоните нам или пришлите сведения с помощью одной из форм обратной связи.
Правила нанесения CE маркировки на медицинские изделия
Регламентами установлены очень четкие правила нанесения СЕ маркировки. Главные требования к CE маркировке приведены в Приложении V и статье 20 Регламента MDR 2017/745:

CE маркировка, наносимая на медицинское изделие
Где получить сертификат СЕ
Для получения CE сертификата просто обратитесь в нашу компанию. Вместе мы подберем нужную схему оформления сертификата, подготовим необходимые документы, проведем испытания и пройдем все проверки со стороны нотифицированных и надзорных органов.
Ответственность за нарушения в сфере обращения сертифицированных в ЕС медицинских изделий
За нарушение правил обращения медицинских изделий в Европейском союзе предусмотрена административная и даже уголовная ответственность. Теоретически возможны следующие ситуации, при которых нарушаются требования Регламентов:
Отличия СЕ сертификации от регистрации в РФ и ЕАЭС
Процедуры регистрации и CE сертификации имеют значительные отличия:
Таким образом, CE сертификация является поэтапным приведением продукта и производителя в соответствие требованиям европейского законодательства. Итогом такого приведения является создание Декларацией соответствия CE и нанесение CE-марки (CE-маркировки).
Наши преимущества
К очевидным плюсам работы с нами можно отнести:
Эти преимущества позволяют свести к минимуму ваше участие, снизить издержки и максимально сократить сроки выхода на европейский рынок.
Вы планируете выводить свои медицинские изделия на Европейский рынок? Тогда свяжитесь с нами любым удобным для вас способом для получения бесплатной консультации. Также вы можете оставить онлайн заявку. Мы рады помочь вам в оформлении CE сертификата и в решении других вопросов, относящихся к сертификации медицинских изделий для экспорта в Европу.




